
For decades, the story of Alzheimer's has been a story of proteins gone wrong. Amyloid beta clumps into plaques. Tau twists into tangles. Neurons die. The tangles, in particular, have been treated as pure wreckage, a sign that a cell's internal scaffolding has collapsed. A new paper argues the tangles might have started as something else entirely: a weapon aimed at a virus.
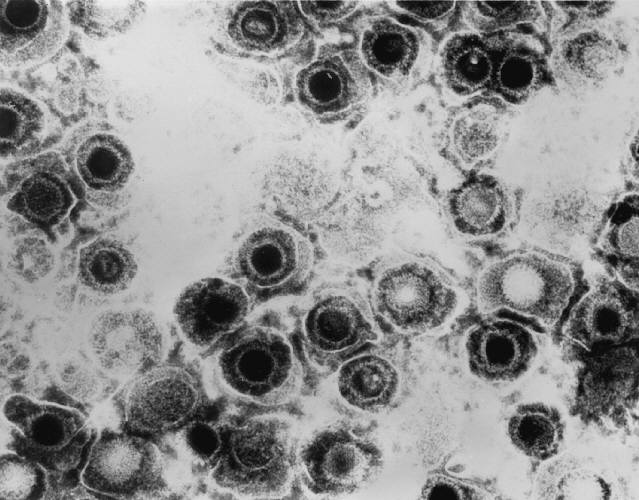
Researchers at Massachusetts General Hospital and Harvard Medical School, led by William A. Eimer with senior author Rudolph E. Tanzi, report in Nature Neuroscience that tau becomes hyperphosphorylated in human neurons when those neurons are infected with a virus. And in that phosphorylated state, tau can neutralize herpes simplex virus 1 (HSV-1) by binding directly to the virus's capsids, the protein shells that hold viral DNA.
Put simply, the protein that fouls up the neuronal cytoskeleton in Alzheimer's appears to be doing something on purpose. It grabs the invader.
From cellular scaffolding to flypaper
Tau normally stabilizes microtubules, the internal tracks that keep a neuron's long processes organized. When tau gets heavily phosphorylated, it falls off those tracks, the microtubules destabilize, and the loose tau aggregates. That cascade is what pathologists see in a diseased brain, and it has long been read as a malfunction.
The Harvard group flips the reading. In their experiments, viral infection was the trigger for the phosphorylation, not a random breakdown. Once modified, tau bound HSV-1 capsids and blocked the virus from establishing infectivity in the neurons. The very features that define "bad" tau in Alzheimer's, the group writes, look like an antiviral response: hyperphosphorylation, microtubules coming apart, and the protein clumping together.
This is not the first time a supposed Alzheimer's culprit has been recast as an immune actor. The same lab previously showed that amyloid beta, the core ingredient of senile plaques, behaves as an antimicrobial peptide that can trap microbes. Now tau joins it. The authors propose that both proteins are part of the brain's innate immune defense, and that plaques and tangles are the aftermath of that defense being deployed.
Why herpes keeps showing up in dementia research
HSV-1 is not a strange choice of virus. It infects most people, hides for life in the nervous system, and periodically reactivates. Epidemiologists have noticed for years that carrying HSV-1 tracks with higher dementia risk in some populations, and antiviral use has been linked to lower risk in others. Those are correlations, and they have been argued about. What this paper offers is a plausible molecular mechanism connecting the two: the virus provokes an antiviral tau response in neurons, and if that response runs often enough or long enough, the byproducts accumulate.
The larger claim is a reframing of the disease itself. If amyloid plaques and neurofibrillary tangles are both products of an orchestrated host defense against microbes, then Alzheimer's neuropathology, alongside the neuroinflammation that accompanies it, may represent an immune program that has overstayed its usefulness. Damage would come not from the defense misfiring once, but from it being called on repeatedly across a lifetime.
What this does not settle
A mechanism in human neurons is not the same as a proven cause of disease in people. The work shows that phosphorylated tau can bind and neutralize a virus; it does not show that this is the dominant driver of tangle formation in every Alzheimer's brain, nor that blocking the virus would prevent dementia. Real brains face many pathogens and stresses, and tau pathology also appears in conditions with no obvious infectious trigger. Whether the antiviral role is central or occasional remains open.
Still, the direction of the argument matters. If tangles begin as protection rather than pure decay, then treatments that simply strip tau away could, in principle, remove a defense the brain was mounting for a reason. The more interesting target might be the trigger, or the failure to shut the response down, rather than the aggregates themselves. That is a different way to think about a disease that has resisted almost every therapy aimed straight at its most famous symptoms.
Comments