
Making a new antibody usually starts with an animal or a person who already has one. You immunize a mouse, or you sort through blood from a recovered patient, and then you fish out the immune cells that happen to make something sticky against your target. It works. It is also slow, expensive, and it fails often. A team at Vanderbilt wanted to skip the biology at the front of that pipeline and let a language model do the first draft instead.
Their tool is called MAGE, short for monoclonal antibody generator. It is a protein language model, the same broad family of software that predicts protein shape from sequence, but fine-tuned for one narrow job: write both chains of a human antibody at once, aimed at a target you specify. The work was published in Cell on November 4, 2025.
Why both chains at once matters
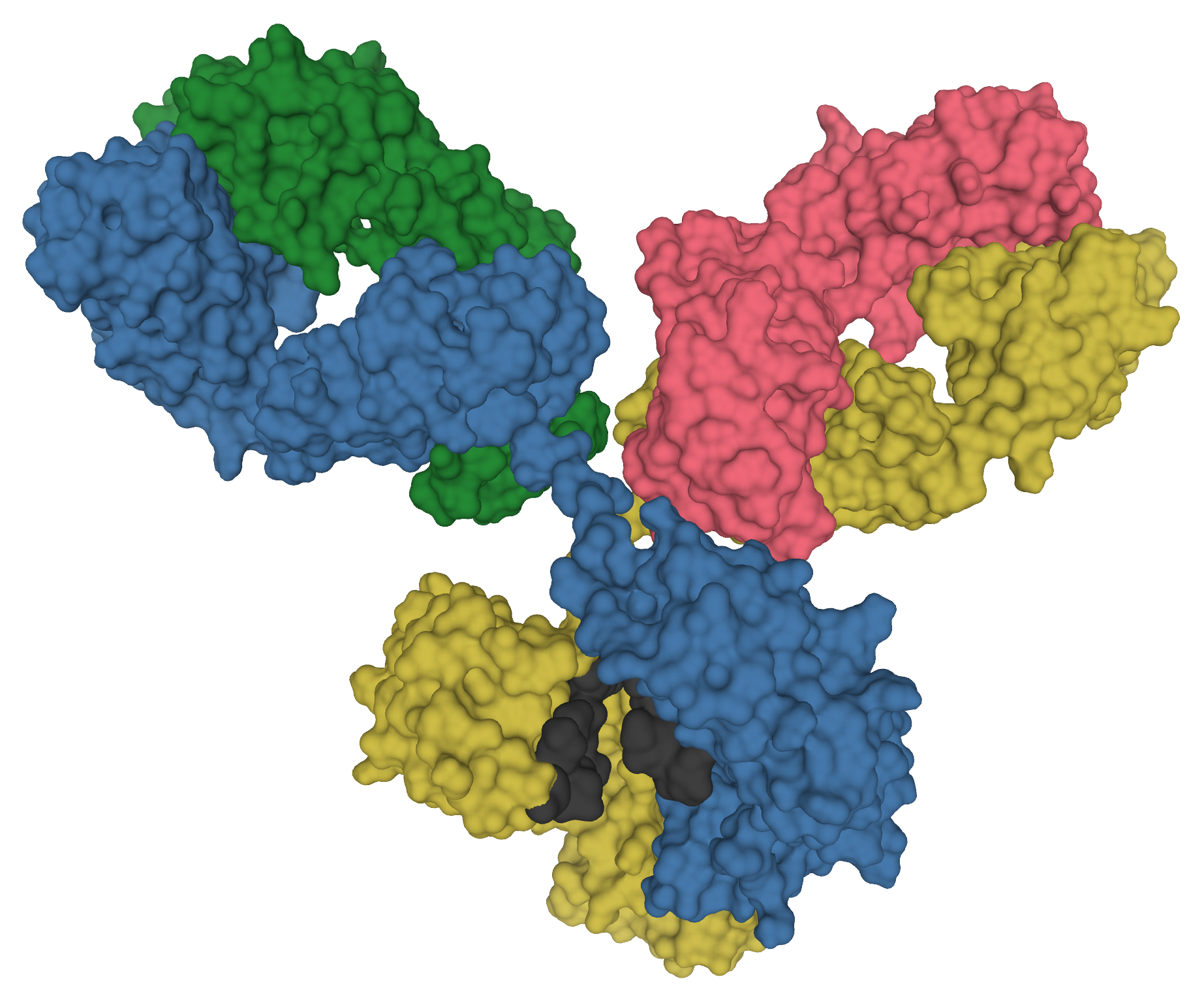
An antibody is not one molecule. It is a pair. A heavy chain and a light chain fold together, and the groove where they meet is what actually grips the target. Most earlier AI approaches optimized a piece of an existing antibody, or generated sequences without any particular target in mind. MAGE does something harder. Given a target, it generates the full variable region of the heavy chain and the light chain together, as a matched set, with no starting template to copy from.
That last part is the claim worth pausing on. The model is not editing a known binder. It is producing candidate antibodies from the target alone. The authors describe MAGE as a first-in-class model for designing human antibodies against multiple targets this way.
Three viruses, real binding data
Software that generates plausible-looking protein sequences is easy to build and easy to fool yourself with. The sequences look fine. Whether they fold, and whether they stick to the thing you care about, is a wet-lab question. So the team made the designs and tested them.
MAGE produced antibodies with confirmed binding against three separate targets: SARS-CoV-2, the virus behind COVID; an H5N1 avian influenza strain, the kind that has been circulating in birds and cattle; and respiratory syncytial virus A, or RSV-A, a common cause of serious infection in infants and older adults. The generated sequences were novel and diverse rather than near-copies of natural antibodies, and the binding specificity was validated experimentally, not just predicted.
Three targets is a small number, but the point is breadth of a different kind. The same model handled all three without being rebuilt for each one. That is the difference between a one-off trick and a general method.
What the paper does not claim
Binding is not the same as neutralizing, and neutralizing in a dish is not the same as protecting a person. A design that grabs onto a viral protein can still fail to block infection, and the abstract stays focused on binding specificity rather than clinical function. Generating sequences is also cheap compared with the screening and characterization that follow, so this shifts the bottleneck rather than removing it. And a model trained on human antibody sequences will reflect whatever is over- or under-represented in that training data, which is worth watching as these tools get pointed at harder or less familiar targets.
There is also the plain fact that antibody discovery has a lot of failure baked in no matter how the first candidates are made. MAGE moves the starting line, not the finish line. Still, moving the starting line is the expensive part. If you can generate a batch of targeted, human, paired-chain candidates from a computer instead of an immunized animal, you change the economics of where a drug program begins. That is the engineering result here, and it is a real one.
Comments